
Abstract.
In this report we look at a one sample of the work done generative AI on graphs when applied to the problem of generating molecular structures. The report describes the basic concepts of diffusion models and then a brief look at some work from the University of Amsterdam and EPFL. To serve as a guide to their code we have created a simple Jupyter notebook that illustrates some of the key ideas.
Introduction
One of the most interesting developments in recent machine learning has been the amazing progress being made in generative AI. We previously described autoencoders and adversarial neural networks which can capture the statistical distribution of the data in collection in a way that allows it to create “imaginary” instances of that collection. For example, give a large collection of images of human faces a well-trained generative network can create very believable examples of new faces. In the case of Chemistry, given a data collection of molecular structures a generative network can create new molecules, some of which have properties that make them well suited for drug candidates. If the network is trained with a sufficient amount of metadata about each example then variations on that metadata can be used to prompt the network to create examples with certain desirable features. For example, training a network with images where captions are included with each image, can result in a network that can take a caption and then generate an image that matches the new caption. Dall.E-2 from OpenAI or Stable-diffusion from Hugging Face do exactly this. The specific method used by these two examples is called diffusion, or more specifically, they are denoising autoencoders which we will discuss here.
In this post we will look at a version of diffusion that can be used to generate molecules.
A Quick Tutorial on Diffusion
The idea behind Diffusion is simple. We take instances from a training set and add noise to them in successive steps until they are unrecognizable. Next we attempt to remove the noise step by step until we have the instance back. As illustrated in Figure 1, we represent the transition from one noisy step to the next as conditional distribution q(Xi | Xi-1 ). Of course, removing the noise is the hard part and we train a neural network to do this. We represent the denoising steps based on that network as pꝋ(Xi-1 | Xi ).
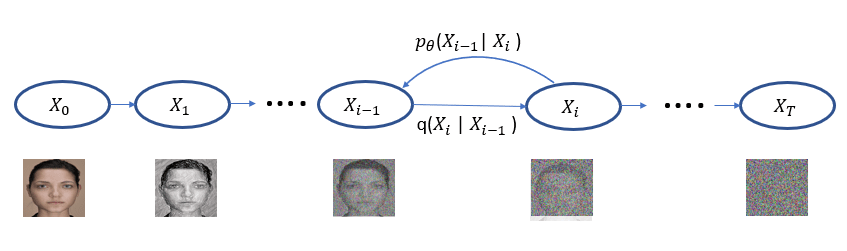
Figure 1. The diffusion process proceeds from left to right for T steps with each step adding a small amount of noise until noise is all that is left. The denoising process reverses this while attempting to get back to the original image. When we have a trained neural network for the denoising step pꝋ defines a distribution that, in the last step, models the distribution of the training data.
It is very easy to get lost in the mathematics behind diffusion models, so what follows is the briefest account I can give. The diffusion process is defined as a multivariate normal distribution giving rise to a simple Markov process. Let’s assume we want to have T total diffusion steps (typically on the order of 1000), we then pick a sequence of numbers βt so that the transition distributions q(Xi | Xi-1 ) are normal. Writing
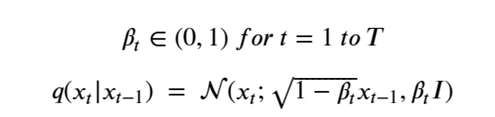
This formulation has the nice property that it can used to compute q(Xi | X0 ) as follows. Let
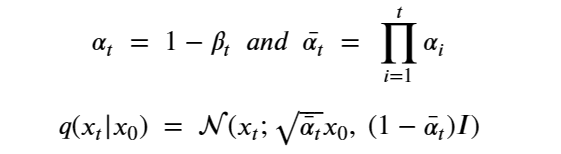
At the risk of confusing the reader we are going to change the variable names so that it will be consistent with the code we will describe in the next sections. We set
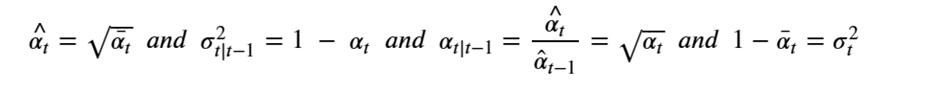
Then, setting X=x0 we can simplify the equation above as
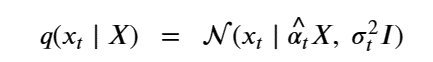
The importance of this is that it is not necessary to step though all the intermediate Xj for j<t to compute Xt. Another way of saying this is
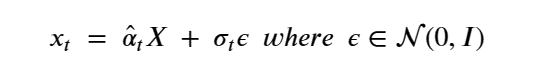
The noise introduced in X to get to xt is σtε. To denoise and compute pꝋ(Xi-1 | Xi ) we introduce and train a network ϕ(xt, t) to model this noise.
After a clever application of Bayes rule and some algebra we can write our denoising step as
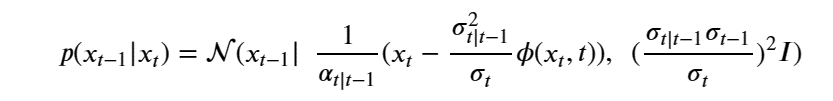
For details see Ho, Jain and Abbeel in Denoising Diffusion Probabilistic Models or the excellent blog by Lilian Weng What are Diffusion Models?.
Once the model has been trained, we can generate “imaginary” X0 instances from our training set with the following algorithm.
Pick XT from N(0, 1)
for t in T..1:
pick ε from N(0,1)
compute
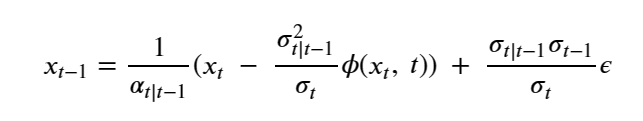
return x0
Figure 2. The denoising algorithm.
This is the basic abstract denoising diffusion algorithm. We next turn to the case of denoising graph networks for molecules.
Denoising Molecular Graphs
An excellent blog post by Michael Galkin describes several interesting papers that present denoising diffusion models for topics related to molecule generation.
The use of deep learning for molecular analysis is extensive. An excellent overview is the on-line book by Andrew D. White “Deep Learning for Molecules & Materials”. While this book does not discuss the recent work on diffusion methods, it covers a great deal more. (I suspect White will get around to this topic in time. The book is still a work in progress.
There are two ways to approach diffusion for molecular structures. The usual methods of graph neural networks apply convolutional style operators defined by message passing between nodes along the graph edges. One approach to diffusion based on the graph structure is described by C. Vignac et. al. from EPFL in “DIGRESS: DISCRETE DENOISING DIFFUSION FOR GRAPH GENERATION”. In the paper they use the discrete graph structure of nodes and edges and stipulate that the noise is applied to each node and each edge independently and governed by a transition matrix Q defined so that for each node x and edge e of a Graph G with X nodes and E edges as
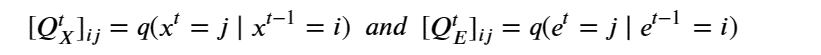
Given a graph as the pair nodes, edges as Gt = (Xt , E t ) the noise transitions is based on matrix multiplications as
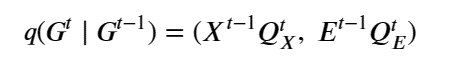
The other approach is to use a representation where the graph is logically complete (every node is “connected” to every other node). The nodes are atom that are described by their 3-D coordinates. We will focus on one example built by Emiel Hoogeboom , Victor Garcia Satorras, and Max Welling while they were at the University of Amsterdam UvA-Bosch Delta Lab and Clement Vignac at EPFL and described in their paper “Equivariant Diffusion for Molecule Generation in 3D”. The code is also available and we will describe many parts of it below.
In studies of this type one of the major datasets used is qm9 (Quantum Machine 9) which provides quantum chemical properties for a relevant, consistent, and comprehensive chemical space of small organic molecules. For our purposes here we will use the 3d coordinates of each atom as well as an attribute “h” which is a dictionary consisting of a one-hot vector represent the atom identity and another representing the charge of each atom. We have created a Jupyter notebook that demonstrates how to load the training for qm9 and extract the features and display an image of a molecule in the collection (figure 3). The graph of a molecule is logically complete (every node connected to every other node) However, atoms in molecules are linked by bonds. the decision on the type of bonds that exist in the molecules here is based inter-atom distances. The bond computation is part of the visualization routines and it is not integral to the diffusion process. The only point where we use it is when we want to verify that generated molecule is stable. (The Jupyter notebook also demonstrates the stability computation.)
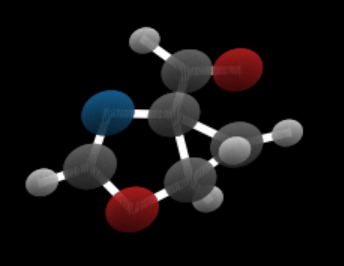
Figure 3. Sample molecule rendering from notebook.
Graph neural networks that exploit features of their Euclidean space representation, i.e. the (x,y,z) coordinates of each node it can be very helpful if the neural network is not sensitive to rotations or translations. This property is called equivariance. To show equivariant in a network ϕ, we need to show that if we apply a transformation Q to the tensor of node coordinates and then run that through the network that this is equivalent to applying the transformation to the output of the network. We keep in mind that the application of the transformation is matrix multiplication and that the node data h is independent of the coordinates. Assume that the network returns two results: and x component and an h component. Then equivariance can be stated as
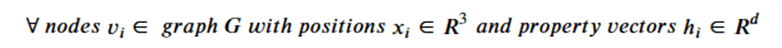
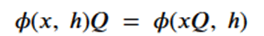
or more formally as
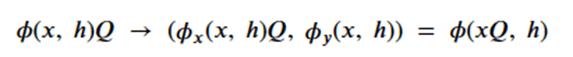
To achieve this, the network (called egnn) computes the sequence,
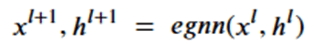
The network is based on 4 subnetworks.
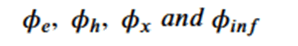
Which are related by the following equations.
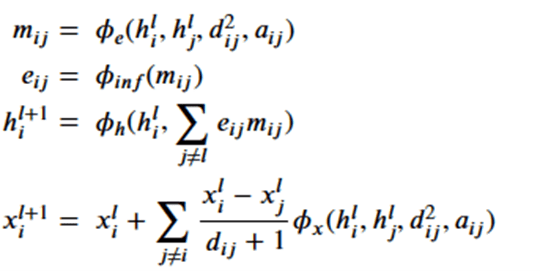
where
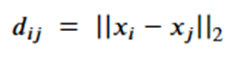
Note that dij is the distance between node I and j and so is invariant to the rotations and translations of the molecule. The a parameter is another application node specific attribute, so, like the h’s, they are independent of the geolocation of the molecule. From this we can conclude the m’s and e’s are also not dependent upon the x’s. That leaves the last equation. Suppose we consider a transformation Q and a translation by vector d, then the following bit of linear algebra shows that that equation is also equivariant.
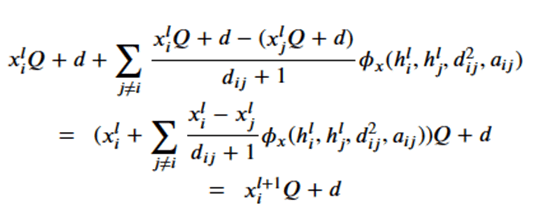
The four subnetworks are assembled into an equivariant block as shown in Figure 4.
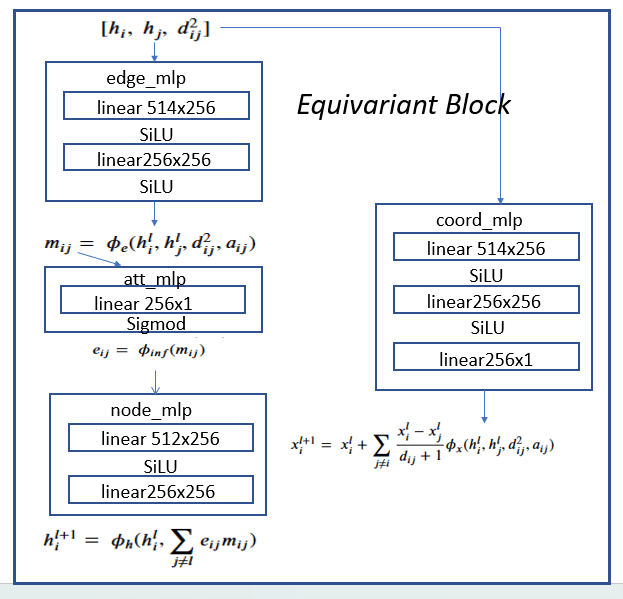
Figure 4. Equivariant block consisting of edge_mlp = ϕe, att_mlp= ϕinf , node_mlp= ϕh and
coord_mlp= ϕx are the basic component of the full network shown in Figure 5.
The h vector at each node of the graph has seven components: a one-hot vector of dimension 5 representing the atom type, an integer representing the atom charge and a float that is associated with the time-step j. The first stage is an embedding of the h vectors in R256. To map it to the edge_mlp requires two h vectors each of length 256 plus the a pair (i, j) representing the edge (this is used to compute the distance between the two node d2ij)
Part 3 of the provided Jupyter Notebook provides an explicit demonstration of the network equivariance.
The full egnn network is shown in Figure 5. It consists of the embedding for h followed by 10 equivariant blocks. These are followed by the output embedding.
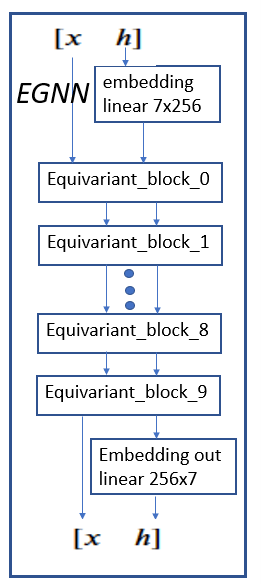
Figure 4. full egnn network
Demonstrating the denoising of molecular noise to a stable molecule
The authors have made it very easy to illustrate the denoising process. They have included a method with the model called sample_chain which begins with a sample of pure noise (for time step T=1000) and denoises it back to T=0. We demonstrate this in the final part of our Jupyter notebook. In the code segment below we have copied the main part of that method.

As you can see this code implements Figure 2. The key function is sample_p_zs_given_zt which contains the code below.

which is the denoising step from figure 2. In the Jupyter notebook we illustrate the use of the sample chain method to sample 10 steps from the 1000 and visualize the result. We repeat the process until the sequence results in a stable molecule. A typical result is shown in Figure 5 below.
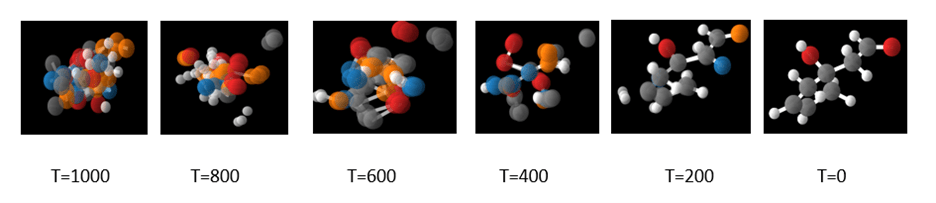
Figure 5. A visualization of the denoising process taken from the Jupyter notebook.
Conclusion
This short paper has been designed as a tour of one example of using diffusion for molecule generation. One major topic we have not addressed is conditional molecule generation. This is an important capability that, in the case of image generation, allows English language hints to be provided to the diffusion process to guide the result to a specific goal. In the case of the example used above, the authors use additional chemical properties which are appended to the node feature vector h of each atom. This is sufficient to allow them to do the conditional generation.
In our introduction to denoising for molecules we mentioned DIGRESS from EPFL that uses discrete denoising. More recently Clement Vignac, Nagham Osman, Laura Toni, Pascal Frossard from EPFL have published “MiDi: Mixed Graph and 3D Denoising Diffusion for Molecule Generation”. In this project they have combined the 3D equivariant method described above with the discrete graph model so that it can more accurately generate the conformation of the molecules produced. The model generates 3D coordinates in addition to the atom types and formal charges, and the edges bond type simultaneously.
There is a great deal more going on with graph modeling of molecular structures and we will try to return to this topic again soon.
